
NAD+ Boosters and Ovarian Aging: Why the Science Just Got More Interesting
A new review reveals how mitochondrial dysfunction drives ovarian aging—and why targeting it might actually work.

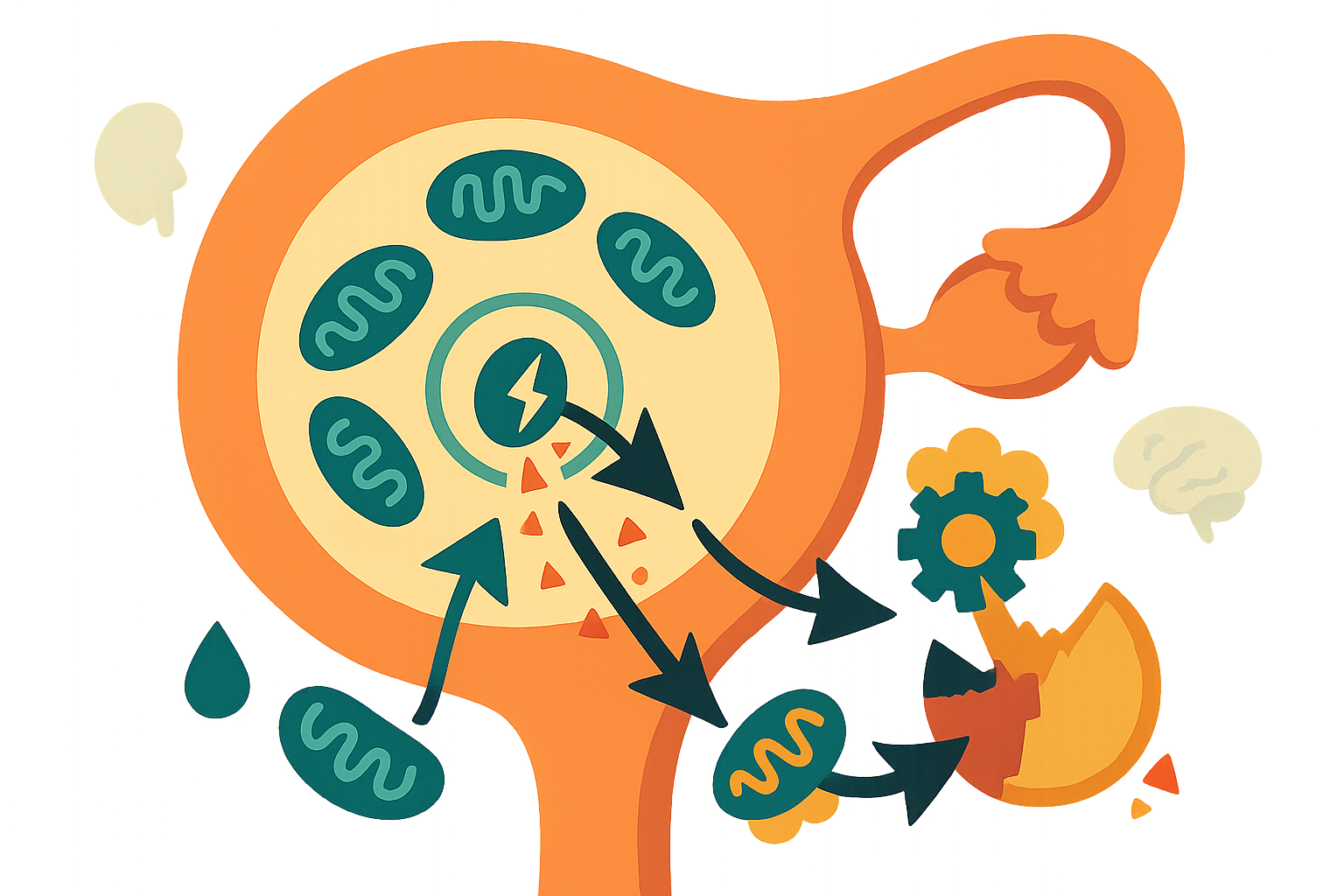
Here’s something that might surprise you: your ovaries age faster than almost any other organ in your body. And it’s not just about running out of eggs. A comprehensive new review in the Journal of Translational Medicine lays out compelling evidence that damaged mitochondria—the cellular powerhouses—trigger a chronic inflammatory cascade that accelerates ovarian decline. The mechanisms identified aren’t set in stone, and several promising interventions are already showing results in preclinical studies.
I’ve experimented with NAD+ boosters in the past—nicotinamide riboside specifically—and frankly, when prices were sky-high, it was hard to justify the expense. But now that costs have dropped significantly, this review has me reconsidering. The potential health benefits seem genuinely wide-ranging, particularly for women concerned about reproductive aging.
“Mitochondrial dysfunction not only represents an early hallmark of ovarian aging but also acts as a key amplifier of chronic inflammation through activation of mitochondria-associated inflammatory pathways.”
What’s the Big Idea?
The research is a detailed mechanistic review connecting mitochondrial breakdown to accelerated ovarian aging. Ovarian aging isn’t just about running out of eggs. It’s driven by a self-perpetuating cycle: damaged mitochondria leak danger signals (mtDAMPs), which trigger inflammatory pathways like NLRP3 and cGAS/STING, creating chronic inflammation that further damages mitochondria and accelerates functional decline.
What caught my attention is how specific this process is. When mitochondria malfunction—through DNA mutations, oxidative stress, or quality control failures—they release molecular fragments into the cell. The immune system recognizes these fragments as threats, launching inflammatory responses originally designed to fight infections. But in aging ovaries, this becomes a vicious cycle. The inflammation damages more mitochondria, which release more danger signals, which drive more inflammation.
The review synthesizes evidence from multiple model systems showing this isn’t just correlation—it’s causation. Genetic deletion of mitochondrial quality control proteins (like MFN2 or PINK1) directly triggers inflammation. Conversely, enhancing mitophagy (the cellular cleanup system for damaged mitochondria) reduces inflammatory signaling and improves oocyte quality.
Why Should You Care?
The implications are substantial for anyone thinking about fertility preservation or managing menopausal health. This framework suggests that ovarian aging might be partially reversible—or at least significantly slowed—by targeting the mitochondria-inflammation axis.
Here’s what makes this practically relevant: The ovary exhibits accelerated aging compared to other organs. It’s one of the earliest systems to show functional decline in humans, typically beginning around age 35 with sharp acceleration afterward. The review argues this isn’t inevitable biological destiny but rather reflects the ovary’s unique vulnerability to mitochondrial stress.
Why? Three reasons: First, oocytes don’t regenerate. Unlike your liver, which can replace damaged cells, eggs you’re born with are all you get. Damaged mitochondria accumulate over decades. Second, the follicular environment becomes an inflammatory echo chamber—granulosa cells and oocytes exchange damaged mitochondria and inflammatory signals through specialized cellular tunnels. Third, declining estrogen levels remove a protective “shield” that normally maintains mitochondrial function and suppresses inflammation.
This creates a feedback loop: mitochondrial damage reduces estrogen production, which removes protective signals that maintain mitochondria, which further reduces estrogen... you get the idea. The review also connects this process to post-menopausal complications—osteoporosis, cardiovascular disease, neurodegeneration—suggesting the inflammation isn’t confined to reproductive tissues.
For women undergoing IVF, there’s direct clinical relevance. Elevated inflammatory markers and mitochondrial DNA in follicular fluid correlate with poorer outcomes. Understanding these mechanisms might lead to better protocols for improving egg quality in older patients or those with diminished ovarian reserve.
What’s Next on the Horizon?
The next frontier is translating these insights into interventions that actually work in humans. Several therapeutic strategies show promise in preclinical models, though we’re still in early stages.
NAD+ precursors (NMN, nicotinamide riboside) are particularly interesting. They activate sirtuins (SIRT1/2), promote mitochondrial biogenesis, and reduce oxidative stress. Multiple mouse studies show improvements in ovarian reserve, oocyte quality, and fertility in aged animals. That’s why I’m personally reconsidering supplementation—the mechanistic rationale is solid, even if human fertility data remains limited.
Mitochondria-targeted antioxidants like MitoQ and SS-31 represent more precise approaches, delivering protective molecules directly to mitochondria. Early results in animal and in vitro studies show improved oocyte maturation and reduced oxidative damage. These compounds address the problem at its source rather than relying on systemic antioxidants with unpredictable tissue distribution.
Perhaps most intriguingly, the review discusses “inflammatory memory”—epigenetic modifications that lock cells into pro-inflammatory states even after the initial trigger resolves. This suggests timing matters enormously. Interventions might be most effective before this memory becomes entrenched, potentially in the early 30s rather than after fertility has already declined noticeably.
Emerging technologies like mitochondrial DNA editing (mito-TALENs) could theoretically correct inherited mutations, though significant safety and efficiency hurdles remain. More immediately practical: metabolic modulators like metformin and L-carnitine show anti-aging effects by improving mitochondrial function and reducing inflammation.
The big unanswered questions revolve around optimal intervention windows, combination therapies, and long-term safety. Many promising compounds work in vitro or in mice, but human ovaries present unique challenges. Who knows, maybe soon we’ll see properly designed clinical trials targeting these pathways specifically for fertility preservation.
Safety, Ethics, and Caveats
The research is honest about significant limitations. Most evidence comes from animal models—primarily mice—where reproductive physiology differs meaningfully from humans. In vitro studies on isolated cells don’t capture the complexity of intact ovarian tissue within living systems.
The inflammatory pathways described are highly conserved and essential for normal immune function. Blocking them too aggressively could have unintended consequences. For instance, NLRP3 inflammasome activation isn’t purely pathological—it plays legitimate roles in ovulation and tissue remodeling. Completely suppressing inflammation might impair normal reproductive processes.
There’s also the question of epigenetic effects. Several studies hint that interventions during critical developmental windows could affect offspring through altered gene expression patterns. Most current research hasn’t rigorously evaluated transgenerational impacts, which seems like a pretty important consideration for fertility treatments.
Safety profiles for many compounds remain incompletely characterized. Mitochondria-targeted therapies are relatively new, and long-term effects in humans are largely unknown. The optimal dose, timing, and duration of treatment remain empirical questions. What works in a 40-week-old mouse might not translate to a 40-year-old woman.
Ethically, there’s tension between individual choice and population-level uncertainty. Should women access experimental mitochondrial therapies based on promising preclinical data, or wait for definitive human trials that might take decades? The review doesn’t address these questions directly, but they’re inherent in any discussion of anti-aging interventions.
From a bias perspective, the review is comprehensive but advocacy-oriented—the authors clearly believe mitochondrial dysfunction is a central driver of ovarian aging and emphasize supportive evidence. Alternative mechanisms (telomere attrition, nuclear DNA damage, cellular senescence) receive less attention. That’s not necessarily wrong, but it’s worth noting the framing.
What This Could Mean for You
The takeaway is that ovarian aging might be more modifiable than previously assumed, particularly through early intervention targeting mitochondrial health.
If you’re in your early-to-mid 30s and concerned about future fertility, lifestyle factors supporting mitochondrial function seem prudent: regular exercise (which enhances mitophagy), avoiding environmental toxins (heavy metals, certain pesticides), managing stress, and maintaining metabolic health. These aren’t revolutionary, but the mechanistic understanding adds weight to recommendations you’ve probably heard before.
For NAD+ supplementation specifically—I’m leaning toward trying nicotinamide riboside again, given the price drops and mechanistic plausibility. The human fertility data isn’t definitive, but preliminary evidence suggests potential benefits for mitochondrial function across multiple systems. Start conservatively (200-400 mg daily based on mouse studies scaled to body weight), and honestly, don’t expect miracles.
Antioxidant supplementation is trickier. General antioxidants like vitamin C or E have mixed evidence. If you’re interested in more targeted approaches, discuss mitochondria-specific options (MitoQ, SS-31) with a knowledgeable physician, though availability and cost may be limiting factors.
If you’re already facing diminished ovarian reserve or undergoing fertility treatments, ask your reproductive endocrinologist about emerging protocols incorporating mitochondrial support. Some clinics are experimenting with CoQ10 supplementation during IVF cycles, for instance, based on evidence it improves oocyte mitochondrial function.
Think about timing. The review suggests mitochondrial dysfunction begins subtly in the early 30s, well before clinical symptoms appear. Waiting until fertility is obviously compromised means working against established inflammatory memory and accumulated damage. Proactive strategies might be more effective than reactive ones.
But—and this is important—don’t let this review induce unnecessary anxiety. Ovarian aging is variable; genetics, lifestyle, and chance all play roles. The mechanisms described are real, but individual trajectories differ substantially. Use this information to make informed choices, not to catastrophize about timelines.
Finally, stay skeptical of supplements marketed specifically for “ovarian health” or “egg quality” without rigorous evidence. The science here is promising but preliminary. Companies are excellent at outrunning the data.
Explore the Full Study
Ju, W., Yan, B., Li, D., Lian, F., & Xiang, S. (2025). Mitochondria-driven inflammation: a new frontier in ovarian aging. Journal of Translational Medicine, 23, 1005. https://doi.org/10.1186/s12967-025-06966-6